The basic question that I try to find an answer to is this:
Is it pure chance that methionine is the universal start amino acid in protein synthesis or is there a logical explanation why this amino acid and not any other (formyl-methionine doesn't count and glutamine or isoleucine are much less efficient at initiation of translation than methionine) is used.
Francis Crick wrote:
"Discussion of the actual amino acids used in the code may not be very profitable. Some less common amino acids, such as cysteine and histidine, would clearly seem to have an advantage because of their chemical reactivity; but whether, say, methionine could be justified in this way seems less obvious."
Crick FH. The origin of the genetic code. J Mol Biol. 1968 Dec;38(3):367-79.
That may very well be true, but I think just opting for the "accident" solution is not a very satisfying answer and to have a logical explanation is better than not having any at all. Of course the structure of the initiator tRNA is the deciding factor in this, but it might have been conceivable that other initiator tRNAs might have been successful. If methionine is nearly always the first amino acid in a protein sequence, but often cleaved off with other N-terminal amino acids, then it might not be essential for the structure/function relation of the protein but still play an important role outside protein synthesis, which is where S-adenosylmethionine (AdoMet) comes in. S-adenosylmethionine is used in transmethylation (of DNA, rRNA, tRNA, mRNA-caps, proteins and lipids), transsulfuration, and aminopropylation (polyamine synthesis of spermidine and spermine). Could the fundamental processes of translation, transmethylation and polyamine synthesis be tethered or is it merely an interesting coincidence, a correlation in the absence of a "cause-and-effect" relation? How could one adress this question? How do you untangle a Gordian knot without using brute force that would wreck the whole system?
Tuesday, August 19, 2008
Friday, August 15, 2008
Mindgames with SAM-analogs
I keep on wondering what different S-adenosylmethionine analogs would do to the cellular metabolism and if such molecules might prove useful for investigating experimental leishmaniasis. Obviously, these molecules are likely to be toxic to the host as well as the parasite, but perhaps there might be a way to limit their exposure to macrophages, targeting amstigotes, and thereby reducing the expected undersired side effects to a minimum.
Compared to S-adenosylmethionine:
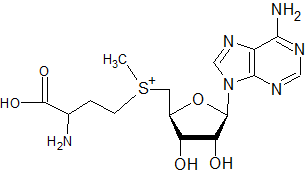
The following molecule would not be expected to act as a substrate in methylation (of DNA, rRNA, mRNA caps, tRNA, proteins, and phospholipids) or the transfer of propylamine, but not interfere with putrescine synthesis directly as sinefungin might:

This one could be able to catalyse the formation of spermidine and spermine, but lead to ethylation instead of methylation:

Whilst this molecule, if accepted by the respective enzymes as a substrate, could only be used for the transfer of propylamine but not for methylation/ethylation:

On the other hand, the following molecule would only act as a substrate for methylation and not for polyamine synthesis:

Whilst tubericidin is a SAM-analog that has been shown to affect L. donovani promastigotes but treatment has also led to tubericidin-resistant strains emerging:

In addition, attention should be paid to the stereoisomeric form of S-adenosylmethionine (AdoMet), the (Sc, Ss) isomer being the biologically active form, the (Sc, Rs) isomer being a potent inhibitor. (See review by Ronald Bentley).
Another idea that crossed my mind was whether methyl donor supplementation during pregnancy might render C57BL/6 or CBA mice more susceptible to leishmaniasis (similar to the results obtained for agouti mice regarding coat colour [Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003 Aug;23(15):5293-300.]).
Compared to S-adenosylmethionine:

The following molecule would not be expected to act as a substrate in methylation (of DNA, rRNA, mRNA caps, tRNA, proteins, and phospholipids) or the transfer of propylamine, but not interfere with putrescine synthesis directly as sinefungin might:

This one could be able to catalyse the formation of spermidine and spermine, but lead to ethylation instead of methylation:

Whilst this molecule, if accepted by the respective enzymes as a substrate, could only be used for the transfer of propylamine but not for methylation/ethylation:

On the other hand, the following molecule would only act as a substrate for methylation and not for polyamine synthesis:

Whilst tubericidin is a SAM-analog that has been shown to affect L. donovani promastigotes but treatment has also led to tubericidin-resistant strains emerging:

In addition, attention should be paid to the stereoisomeric form of S-adenosylmethionine (AdoMet), the (Sc, Ss) isomer being the biologically active form, the (Sc, Rs) isomer being a potent inhibitor. (See review by Ronald Bentley).
Another idea that crossed my mind was whether methyl donor supplementation during pregnancy might render C57BL/6 or CBA mice more susceptible to leishmaniasis (similar to the results obtained for agouti mice regarding coat colour [Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003 Aug;23(15):5293-300.]).
Sunday, August 10, 2008
S-adenosylmethionine and sinefungin
There is always the possibility that I am only seeing what I want to see, which is in itself slightly unnerving and the reason why I repeatedly ask for feedback from anyone out there. Whether or not anyone who stumbles across these pages is actually involved in science and could potentially use these ideas is another matter. For the time being, all I can do is to write down my thoughts and hope that some day someone will know what to do with them.
I recently came across an old paper on antileishmanial properties of the S-adenosylmethionine-analog sinefungin:
Nolan LL. Molecular target of the antileishmanial action of sinefungin. Antimicrob Agents Chemother. 1987 Oct;31(10):1542-8.
We'll see if and how things may fit together, but sinefungin-mediated inhibition of polyamine synthesis or DNA methylation/ epigenetics might be possible mechanisms worth considering... In fact, sinefungin looks especially cool because it combines being a SAM/SAH-homologue as well as an ornithine derivative...
S-adenosylmethionine

(taken from "http://www.freepatentsonline.com/7048948-0-large.jpg")
Sinefungin

(taken from "http://www.sigmaaldrich.com/thumb/structureimages/59/s______s8559.gif")
Leishmania donovani was shown to be very sensitive to treatment with sinefungin, with even promastigotes being affected by the treatment. Which makes me start to wonder what might have happened to sinefungin in the pipeline to becoming a widely used antileishmanial drug? Is it possible to devise a delivery method for sinefungin which would target macrophages specifically, either by liposomes or linked to peptides that bind to macrophage receptors?
Bachrach U, Schnur LF, El-On J, Greenblatt CL, Pearlman E, Robert-Gero M, Lederer E. Inhibitory activity of sinefungin and SIBA (5'-deoxy-5'-S-isobutylthio-adenosine) on the growth of promastigotes and amastigotes of different species of Leishmania. FEBS Lett. 1980 Dec 1;121(2):287-91.
Phelouzat MA, Basselin M, Lawrence F, Robert-Gero M. Sinefungin shares AdoMet-uptake system to enter Leishmania donovani promastigotes. Biochem J. 1995 Jan 1;305 (Pt 1):133-7.
I recently came across an old paper on antileishmanial properties of the S-adenosylmethionine-analog sinefungin:
Nolan LL. Molecular target of the antileishmanial action of sinefungin. Antimicrob Agents Chemother. 1987 Oct;31(10):1542-8.
We'll see if and how things may fit together, but sinefungin-mediated inhibition of polyamine synthesis or DNA methylation/ epigenetics might be possible mechanisms worth considering... In fact, sinefungin looks especially cool because it combines being a SAM/SAH-homologue as well as an ornithine derivative...
S-adenosylmethionine

(taken from "http://www.freepatentsonline.com/7048948-0-large.jpg")
Sinefungin

(taken from "http://www.sigmaaldrich.com/thumb/structureimages/59/s______s8559.gif")
Leishmania donovani was shown to be very sensitive to treatment with sinefungin, with even promastigotes being affected by the treatment. Which makes me start to wonder what might have happened to sinefungin in the pipeline to becoming a widely used antileishmanial drug? Is it possible to devise a delivery method for sinefungin which would target macrophages specifically, either by liposomes or linked to peptides that bind to macrophage receptors?
Bachrach U, Schnur LF, El-On J, Greenblatt CL, Pearlman E, Robert-Gero M, Lederer E. Inhibitory activity of sinefungin and SIBA (5'-deoxy-5'-S-isobutylthio-adenosine) on the growth of promastigotes and amastigotes of different species of Leishmania. FEBS Lett. 1980 Dec 1;121(2):287-91.
Phelouzat MA, Basselin M, Lawrence F, Robert-Gero M. Sinefungin shares AdoMet-uptake system to enter Leishmania donovani promastigotes. Biochem J. 1995 Jan 1;305 (Pt 1):133-7.
Thursday, August 07, 2008
This is a tRNA's world, but it would nothing, not one little thing, without an amino acid or a peptide...
With the print version of my contribution to the Journal of Theoretical Biology made available today, here's a question that has been on my mind for quite some time.
I keep on wondering, in what way the basic components of the genetic code may have evolved in the absence of any translational machinery. In other words, is it possible that precursors to today's tRNAs simply bound to amino acids thus enabling them to accumulate in higher concentrations than would have been possible in the absence of such interactions? tRNA-like molecules that bound to hydrophobic amino acids may have been able to interact with lipids at the liquid/air or at the liquid/solid interphase. With an accumulation of those tRNA-like molecules other tRNA-like molecules that bound to hydrophilic amino acids may have in turn formed aggregates with the hydrophobic amino acid-binding tRNAs via the codon-equivalent regions, leading to the basic dichotomy between N-U-N and N'-A-N' codons and to a microenvironment with favourable conditions for interactions between different species of nucleic and amino acids. The hydrophilic amino acids would have to balance the charge inequalities leading to basic and acidic amino acids landing close codon proximity. The next step would be that RNA molecules with catalytic activity bound these adaptor RNAs at strategic positions and the attached amino acids started to be a part if the catalytic process. Finally, the translational machinery would have evolved and the code would have continued to change with it. Some RNA-amino acid pairings would have been thrown out of the race and new ones joined the process at this stage. A drive to reduce codon ambiguity and to minimize errors in translation are likely to be two important factors in the evolution of the code, but some initial rules laid down by the pre-translation system were already in place before the era of large-scale protein synthesis.
I am thankful for any comments you might care to send my way.
If you are interested in more literature on the genetic code, I can recommend:
Nirenberg, MW, Matthaei, JH, Jones, OW. An intermediate in the biosynthesis of polyphenylalanine directed by synthetic template RNA. Proc Natl Acad Sci U S A. 1962 Jan 15;48:104-9.
Woese, CR. On the evolution of the genetic code. Proc Natl Acad Sci U S A. 1965 Dec;54(6):1546-52.
Crick, FH. The origin of the genetic code. J Mol Biol. 1968 Dec;38(3):367-79.
Orgel, LE. Evolution of the genetic apparatus. J Mol Biol. 1968 Dec;38(3):381-93.
Wong, JT. A co-evolution theory of the genetic code. Proc Natl Acad Sci U S A. 1975 May;72(5):1909-12.
Taylor, FJ, Coates, D. The code within the codons. Biosystems. 1989;22(3):177-87.
Szathmáry, E. The origin of the genetic code: amino acids as cofactors in an RNA world. Trends in Genetics. 1999 June;15(6): 223-9.
Massey, SE. A sequential "2-1-3" model of genetic code evolution that explains codon constraints. J Mol Evol. 2006 Jun;62(6):809-10. Epub 2006 Apr 11.
I keep on wondering, in what way the basic components of the genetic code may have evolved in the absence of any translational machinery. In other words, is it possible that precursors to today's tRNAs simply bound to amino acids thus enabling them to accumulate in higher concentrations than would have been possible in the absence of such interactions? tRNA-like molecules that bound to hydrophobic amino acids may have been able to interact with lipids at the liquid/air or at the liquid/solid interphase. With an accumulation of those tRNA-like molecules other tRNA-like molecules that bound to hydrophilic amino acids may have in turn formed aggregates with the hydrophobic amino acid-binding tRNAs via the codon-equivalent regions, leading to the basic dichotomy between N-U-N and N'-A-N' codons and to a microenvironment with favourable conditions for interactions between different species of nucleic and amino acids. The hydrophilic amino acids would have to balance the charge inequalities leading to basic and acidic amino acids landing close codon proximity. The next step would be that RNA molecules with catalytic activity bound these adaptor RNAs at strategic positions and the attached amino acids started to be a part if the catalytic process. Finally, the translational machinery would have evolved and the code would have continued to change with it. Some RNA-amino acid pairings would have been thrown out of the race and new ones joined the process at this stage. A drive to reduce codon ambiguity and to minimize errors in translation are likely to be two important factors in the evolution of the code, but some initial rules laid down by the pre-translation system were already in place before the era of large-scale protein synthesis.
I am thankful for any comments you might care to send my way.
If you are interested in more literature on the genetic code, I can recommend:
Nirenberg, MW, Matthaei, JH, Jones, OW. An intermediate in the biosynthesis of polyphenylalanine directed by synthetic template RNA. Proc Natl Acad Sci U S A. 1962 Jan 15;48:104-9.
Woese, CR. On the evolution of the genetic code. Proc Natl Acad Sci U S A. 1965 Dec;54(6):1546-52.
Crick, FH. The origin of the genetic code. J Mol Biol. 1968 Dec;38(3):367-79.
Orgel, LE. Evolution of the genetic apparatus. J Mol Biol. 1968 Dec;38(3):381-93.
Wong, JT. A co-evolution theory of the genetic code. Proc Natl Acad Sci U S A. 1975 May;72(5):1909-12.
Taylor, FJ, Coates, D. The code within the codons. Biosystems. 1989;22(3):177-87.
Szathmáry, E. The origin of the genetic code: amino acids as cofactors in an RNA world. Trends in Genetics. 1999 June;15(6): 223-9.
Massey, SE. A sequential "2-1-3" model of genetic code evolution that explains codon constraints. J Mol Evol. 2006 Jun;62(6):809-10. Epub 2006 Apr 11.
Thursday, July 31, 2008
SOCS3 expression in classically activated rat macrophages impairs arginase expression
Well, after nearly dismissing my ideas on SOCS and leishmaniasis in my last post, I came across this interesting paper by Liu and colleagues that rekindled the old flame a bit. Published in the Journal of Immunology in May 2008, they show that SOCS3 knockdown by RNA interference in classically activated rat macrophages coincided with an upregulation of alternative activation makers such as arginase.
Liu Y, Stewart KN, Bishop E, Marek CJ, Kluth DC, Rees AJ, Wilson HM. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and in vivo. J Immunol. 2008 May 1;180(9):6270-8.

This brings me back to my question: would SOCS3 shut down STAT3 (IL-6-induced arginase) and IRS-2 signalling and SOCS5 shut down STAT6 (IL-4-induced arginase) signalling so that arginase activity would drop to levels below those able to support parasite proliferation? What role does S-adenosylmethionine play in all this - control of SOCS expression via DNA methylation versus polyamine synthesis of spermine and spermidine? Untangling all this experimentally will no doubt be a bit tricky, because the effects of SOCS on macrophages should be kept separate from effects of SOCS on other cell types.
I would suggest to start in vitro and to compare the effects of RNAi mediated by siRNA against SOCS1, SOCS3, SOCS5, DNA (cytosine-5-)-methyltransferase 1, DNA (cytosine-5-)-methyltransferase 3 alpha, DNA (cytosine-5-)-methyltransferase 3 beta, S-adenosylmethionine synthetase, ornithine decarboxylase, S-adenosylmethionine decarboxylase, spermidine synthase and spermine synthase in respect to parasite development. An interesting side project would be to find out whether or not culturing macrophages in the presence of increased S-adenosylmethionine concentrations leads to higher parasite proliferation. The in vivo experiments that spring to mind are inducible macrophage-specific expression of transgenes: either SOCS-specific shRNA in resistant mice or SOCS in susceptible mice. Whilst it would be preferable if the gene expression was not leaky, I think my wish list is getting slightly too demanding, but if anyone out there thinks that some of these ideas are useful and has the means to set up the necessary experiments please feel welcome to do so. I believe that the sooner the scientific community can come up with new affordable drugs against this neglected tropical disease the better. I would argue the best way to achieve this is through cooperation rather than competition. As always, feedback is greatly appreciated.
Liu Y, Stewart KN, Bishop E, Marek CJ, Kluth DC, Rees AJ, Wilson HM. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and in vivo. J Immunol. 2008 May 1;180(9):6270-8.

This brings me back to my question: would SOCS3 shut down STAT3 (IL-6-induced arginase) and IRS-2 signalling and SOCS5 shut down STAT6 (IL-4-induced arginase) signalling so that arginase activity would drop to levels below those able to support parasite proliferation? What role does S-adenosylmethionine play in all this - control of SOCS expression via DNA methylation versus polyamine synthesis of spermine and spermidine? Untangling all this experimentally will no doubt be a bit tricky, because the effects of SOCS on macrophages should be kept separate from effects of SOCS on other cell types.
I would suggest to start in vitro and to compare the effects of RNAi mediated by siRNA against SOCS1, SOCS3, SOCS5, DNA (cytosine-5-)-methyltransferase 1, DNA (cytosine-5-)-methyltransferase 3 alpha, DNA (cytosine-5-)-methyltransferase 3 beta, S-adenosylmethionine synthetase, ornithine decarboxylase, S-adenosylmethionine decarboxylase, spermidine synthase and spermine synthase in respect to parasite development. An interesting side project would be to find out whether or not culturing macrophages in the presence of increased S-adenosylmethionine concentrations leads to higher parasite proliferation. The in vivo experiments that spring to mind are inducible macrophage-specific expression of transgenes: either SOCS-specific shRNA in resistant mice or SOCS in susceptible mice. Whilst it would be preferable if the gene expression was not leaky, I think my wish list is getting slightly too demanding, but if anyone out there thinks that some of these ideas are useful and has the means to set up the necessary experiments please feel welcome to do so. I believe that the sooner the scientific community can come up with new affordable drugs against this neglected tropical disease the better. I would argue the best way to achieve this is through cooperation rather than competition. As always, feedback is greatly appreciated.
Monday, May 26, 2008
Arginase and ageing - rethinking SAM
Age-Related Alteration of Arginase Activity Impacts on Severity of Leishmaniasis
is a very exciting paper, so convincing in fact that it makes me question whether my hypothesis on SAM is of any use at all to explain susceptibility to leishmaniasis. Arginase activity has been shown time and time again to be crucial to parasite proliferation, but I always wondered whether there was some proverbial elephant standing in the room that no-one mentioned. The SAM story was meant to complement and expand the arginase part and I still believe that with its dual role in polyamine synthesis and regulation of gene expression amongst others, SAM might still be shown to play a role in this experimental system.
At first, I thought that by changes in DNA methylation SOCS expression would be altered affecting cell signalling and arginase activity in host macrophages. This may still be the case, but maybe it would be better to focus on the polyamine synthesis aspect at first. If antimony treatment should deplete SAM levels this could be investigated by incubating macrophage cultures with antimony and measure polyamine levels by thin layer chromatography as carried out by Modolell and colleagues.

is a very exciting paper, so convincing in fact that it makes me question whether my hypothesis on SAM is of any use at all to explain susceptibility to leishmaniasis. Arginase activity has been shown time and time again to be crucial to parasite proliferation, but I always wondered whether there was some proverbial elephant standing in the room that no-one mentioned. The SAM story was meant to complement and expand the arginase part and I still believe that with its dual role in polyamine synthesis and regulation of gene expression amongst others, SAM might still be shown to play a role in this experimental system.
At first, I thought that by changes in DNA methylation SOCS expression would be altered affecting cell signalling and arginase activity in host macrophages. This may still be the case, but maybe it would be better to focus on the polyamine synthesis aspect at first. If antimony treatment should deplete SAM levels this could be investigated by incubating macrophage cultures with antimony and measure polyamine levels by thin layer chromatography as carried out by Modolell and colleagues.

Friday, May 09, 2008
Letting go - now back to Leishmania
Letting go is hard to do, especially when you have fought tooth and nail to get your point across and went through some crazy times. I recently attended the "God Be in My Head" event at the Dana Centre, where a panel chaired by Colin Blakemore discussed the possibility of so-called "God spots" in the brain, and although epilepsy and schizzophrenia were mentioned as possible links to abstract thinking, creativity and spiritual experience, I was surprised to observe that no-one at the event mentioned mania, which seems to me to lie at the intersection between the two other states of mind. One of the best suggestions of the evening was the idea to analyse the brain activity of problem-solving scientists using functional MRI and to try to compare the results to the brain scans of nuns praying or monks meditating. I like it even though I am getting a bit tired of functional MRI papers, but somehow I don't see that experiment being carried out any time soon, the eureka experience and phenomena such as calculation-induced halucinations are science's dirty little secrets and I doubt whether scientists really are prepared to address these issues.
At any rate, I'm digressing, whilst it is fun to rejoin the scientific debate and to watch my representation of the genetic code climb the "google charts", I have to let go of certain ideas and redirect my focus on my PhD. Consequently, I find myself thinking more and more about Leishmania these days. Given that S-adenosyl methionine (SAM) is involved in a number of different cellular processes possibly affecting the replication of intracellular parasites, how would it be possible to differentiate the effects of the individual processes? The ones I am particularly interested in are polyamine synthesis and DNA methylation. Would it be enough to add radiolabelled SAM to infected cells and to try to see where the marked methyl group might end up? Is there radiolabelled SAM available in which the non-methyl carbon atoms are 14C, making the substance useful in tracking polyamines? Whilst I would try to focus on in vitro experiments at first keeping things as simple as possible, I also wonder what in vivo experiments might look like. What effect might adding or depleting SAM have in vivo? What about methylthioadenosine (MTA), a product of the SAM catabolism that arises after the polyamine synthesis step? How could the effect of SAM derivatives be untangled from possible changes to cytokine production?
Too many questions for me to answer...
At any rate, I'm digressing, whilst it is fun to rejoin the scientific debate and to watch my representation of the genetic code climb the "google charts", I have to let go of certain ideas and redirect my focus on my PhD. Consequently, I find myself thinking more and more about Leishmania these days. Given that S-adenosyl methionine (SAM) is involved in a number of different cellular processes possibly affecting the replication of intracellular parasites, how would it be possible to differentiate the effects of the individual processes? The ones I am particularly interested in are polyamine synthesis and DNA methylation. Would it be enough to add radiolabelled SAM to infected cells and to try to see where the marked methyl group might end up? Is there radiolabelled SAM available in which the non-methyl carbon atoms are 14C, making the substance useful in tracking polyamines? Whilst I would try to focus on in vitro experiments at first keeping things as simple as possible, I also wonder what in vivo experiments might look like. What effect might adding or depleting SAM have in vivo? What about methylthioadenosine (MTA), a product of the SAM catabolism that arises after the polyamine synthesis step? How could the effect of SAM derivatives be untangled from possible changes to cytokine production?
Too many questions for me to answer...
Friday, May 02, 2008
Nearly there...
Finally, the "Journal of Theoretical Biology" accepted my manuscript for publication as a letter to the editor. The unedited version of the article can be found ahead of print online at
http://dx.doi.org/10.1016/j.jtbi.2008.04.028.
What a relief. I know it's just one little paper and the impact factor of the journal could be higher, but for some reason this paper is incredibly important to me. I don't know if I have made a huge fool of myself or not, but it's too late anyway. I tried to keep the article as short as possible and did not include any acknowledgements, but of course I would not have been able to persevere in this ridiculous struggle had I not had the support of some incredible people along the way. I don't know whether mRNA-tRNA interaction occurs in a 2-1-2-3 way, but I find the sheer oddness of a language that starts with the middle instead of the beginning of an information unit exciting and cannot get the picture of tRNA-precursor molecules forming aggregates with the second codon base acting as an anchor out of my head. Imagine the message
"hteatsiksontosmcuhotseewahtonoenhsayteseenubtottihnwkhtanoonheaysetthuogthaobutthtawihcehvreyobdsyese."
being translated as "thetaskisnotsomuchtoseewhatnoonehasyetseenbuttothinkwhatnoonehasyetthoughtaboutthatwhicheverybodysees." and finally for the information to "fold" into the beautiful quote: "The task is not so much to see what no one has yet seen but to think what no one has yet thought, about that which everybody sees."
Of course if the interaction between mRNA and tRNA occurred in a 1-2-3-way, the importance of the second base might be explained by a longer interval spent reading the base, so the order would roughly be 1-2-2-3. If the binding of codon and anticodon happened in such a way that all three bases bound simultaneously, then the base in the middle might spend the longest time bound to its cognate because the bases 5' and 3' of it would act as a sort of buffer (like velcro, the middle bit usually the hardest to get off first).
With possible closure on the rearrangement of the genetic code in sight, I wonder if somebody else might pick up on the methionine story. Perhaps one group might try to engineer an organism that uses an initiator-tRNA charged with isoleucine, valine, threonine or homocysteine instead of methionine and report on what the resulting phenotype might be...
But all of this has merely been a side project, the biggest reward for me would be if my initial hypothesis about the involvement of S-adenosylmethionine in determining resistance or susceptibility in experimental leishmaniasis, might prove to be useful.
http://dx.doi.org/10.1016/j.jtbi.2008.04.028.
What a relief. I know it's just one little paper and the impact factor of the journal could be higher, but for some reason this paper is incredibly important to me. I don't know if I have made a huge fool of myself or not, but it's too late anyway. I tried to keep the article as short as possible and did not include any acknowledgements, but of course I would not have been able to persevere in this ridiculous struggle had I not had the support of some incredible people along the way. I don't know whether mRNA-tRNA interaction occurs in a 2-1-2-3 way, but I find the sheer oddness of a language that starts with the middle instead of the beginning of an information unit exciting and cannot get the picture of tRNA-precursor molecules forming aggregates with the second codon base acting as an anchor out of my head. Imagine the message
"hteatsiksontosmcuhotseewahtonoenhsayteseenubtottihnwkhtanoonheaysetthuogthaobutthtawihcehvreyobdsyese."
being translated as "thetaskisnotsomuchtoseewhatnoonehasyetseenbuttothinkwhatnoonehasyetthoughtaboutthatwhicheverybodysees." and finally for the information to "fold" into the beautiful quote: "The task is not so much to see what no one has yet seen but to think what no one has yet thought, about that which everybody sees."
Of course if the interaction between mRNA and tRNA occurred in a 1-2-3-way, the importance of the second base might be explained by a longer interval spent reading the base, so the order would roughly be 1-2-2-3. If the binding of codon and anticodon happened in such a way that all three bases bound simultaneously, then the base in the middle might spend the longest time bound to its cognate because the bases 5' and 3' of it would act as a sort of buffer (like velcro, the middle bit usually the hardest to get off first).
With possible closure on the rearrangement of the genetic code in sight, I wonder if somebody else might pick up on the methionine story. Perhaps one group might try to engineer an organism that uses an initiator-tRNA charged with isoleucine, valine, threonine or homocysteine instead of methionine and report on what the resulting phenotype might be...
But all of this has merely been a side project, the biggest reward for me would be if my initial hypothesis about the involvement of S-adenosylmethionine in determining resistance or susceptibility in experimental leishmaniasis, might prove to be useful.
Sunday, April 27, 2008
Early nerdcore
Over at the excellent www.biology-blog.com I came across this wonderful video mixing free love and protein synthesis (trippy translation). I want more of this stuff: "The Knife" in charge of TLR signalling, "Roisin Murphy" could sing about MHC class I presentation whilst "Hot Chip" serenade MHC class II, cathepsins and lysosomes and "Portishead" would present apoptosis like no-one has ever done before...
Science classes would never be the same again...
Here's a brilliant clip I came across in 2009: Carl Sagan rocks!
Wednesday, November 14, 2007
SAM and polyamines
Just to emphasise that methylation is not the only trick up SAM's sleeve, it's also involved in the production of the polyamines. Spermidine and spermine are synthesised using the putrescine backbone derived from arginine:
arginine -(arginase)-> ornithine -(ornithine decarboxylase)-> putrescine
S-adenosyl methionine -(S-adenosyl methionine decarboxylase)-> decarboxylated SAM
putrescine + decarboxylated SAM -(spermidine synthetase)-> spermidine
spermidine + decarboxylated SAM -(spermine synthetase)-> spermine
Polyamines are important for balancing the negative charge of nucleic acids amongst other things and are therefore essential in all cells and especially in cells undergoing proliferation. For instance, in my preferred animal model, polyamines have been shown to be key factors controlling Leishmania major proliferation.

arginine -(arginase)-> ornithine -(ornithine decarboxylase)-> putrescine
S-adenosyl methionine -(S-adenosyl methionine decarboxylase)-> decarboxylated SAM
putrescine + decarboxylated SAM -(spermidine synthetase)-> spermidine
spermidine + decarboxylated SAM -(spermine synthetase)-> spermine
Polyamines are important for balancing the negative charge of nucleic acids amongst other things and are therefore essential in all cells and especially in cells undergoing proliferation. For instance, in my preferred animal model, polyamines have been shown to be key factors controlling Leishmania major proliferation.

Sunday, September 23, 2007
Selenocysteine and Pyrrolysine
More a postscriptum than a real post, but isn't it amazing, how selenocysteine and pyrrolysine, the 21st and 22nd proteinaceous amino acids, fit into the 2-1-3 genetic code scheme?
Selenocysteine (Sec / U): mRNA codon UGA. Related to cysteine, is in the same subgroup as tryptophane and cysteine and is in the same group as serine.
Pyrrolysine (Pyl / O): mRNA codon UAG. Consisting of a lysine backbone and a pyrrol ring containing a pi electron pair just as in tyrosine, histidine, tryptophane and phenylalanine, is in the same subgroup as tyrosine, and in the same group as histidine and lysine.
It's almost spooky, don't you think?
Selenocysteine (Sec / U): mRNA codon UGA. Related to cysteine, is in the same subgroup as tryptophane and cysteine and is in the same group as serine.
Pyrrolysine (Pyl / O): mRNA codon UAG. Consisting of a lysine backbone and a pyrrol ring containing a pi electron pair just as in tyrosine, histidine, tryptophane and phenylalanine, is in the same subgroup as tyrosine, and in the same group as histidine and lysine.
It's almost spooky, don't you think?
Monday, September 10, 2007
A number of genetic code diagrams
Bresch and Hausmann took Crick's matrix table of the genetic code, i.e. the decoding instructions of translation in which the information stored in the sequence of nucleic acids is transferred to the sequence of amino acids, and were the first to publish a circular diagram of the code.

However, the arrangements of codon bases is in some way arbitrary. The repetitive motif UCAG separates bases according to size (pyrimidine bases U and C, purine bases A and G fall together), groups the inosine-binding bases U, C and A together and allows for the amino acids methionine and isoleucine to be listed in separate groups. When adhering to a constant string of bases, the UCAG motif offers a higher degree of packing of the code as demonstrated by Serguei Lenski on his homepage, which contains a mathematical approach to packing of the genetic code. His results indicate that listing codons in the order of 2-1-3 and retaining the UCAG motif offers an optimal amount of packing, but that does not mean that there aren't any other ways to represent the code. The physicist Yurij Rumer (1901-1985) preferred the motif CGUA for various reasons, placing more emphasis on the strength of bonds formed between the cognate bases (C-G forming three hydrogen bonds, A-U forming only two, see D. A. Semenov's paper if you have no access Rumer's original publication in Russian: Rumer IuB. [Codon systematization in the genetic code] Dokl Akad Nauk SSSR. 1966 Apr 21;167(6):1393-4.).
I believe that the motif AGCU (or UCGA) when viewed from a circular perspective offers a happy compromise between the two. Furthermore, by placing the second codon base at the centre of the diagram it becomes possible to unite the codons of leucine, serine, arginine and stop and to see codons cluster into groups according to the chemical properties of the respective amino acid: M, I, V, L, F are all hydrophobic; K, N, D, E, Q, H, Y are all hydrophilic, T, S and hydoxy-proline carry hydroxyl groups whilst A is structurally related to S; the last group comprises amino acids at the extremes from the smallest G to the largest W, the most hydrophilic R to the hydrophobe C, however S is structurally related to C and C, U, S and G can be converted into one another biosynthetically. In addition, S is a substrate for W synthesis (for more information on the role of biosynthetic pathways in shaping the genetic code, I would recommend: Wong JT. A co-evolution theory of the genetic code. Proc Natl Acad Sci U S A. 1975 May;72(5):1909-12.). Finally, the rare genetically encoded amino acid pyrrolysine (O) found in members of the Methanosarcinaceae family of archeaea is a lysine derivative contianing a pyrrol ring, which reminds me of the structures found in the amino acids W, Y and H and somehow fits neatly into the scheme with proximity to K, H, Y and W.

The 3D model of the genetic code along 2-1-3 rules allows a number of projections that represent distorted "maps" of the code. In this post are three projections of the standard code as well as four diagrams for variants of the standard code, found in mitochondria, mycoplasma, cilliates and green algae, although these graphs are oversimplified. For the recently evolved genetic code in Candida where an L codon has changed into an S codon it is not possible to combine the codons in a way that allows for all codons to be grouped together according to their respective amino acid. Even so, one can follow the small changes that make such a big difference, with the bases 1 and 2 both remaining pyrimidines in the genetic code of Candida. For details of these and more variants of the standard code I would like to draw your attention to the taxonomy browser at NCBI. For more of my thoughts on the genetic code you can follow this link to blog.rna-game.org. Thank you.
Standard code: 1

Standard code: 2

Standard code: 3

Vertebrate Mitochondria Code

Invertebrate Mitochondria Code

Yeast Mitochonria Code
(Codons CGA and CGC absent)

Mycoplasma

Cilliates and green algae

Cilliates


However, the arrangements of codon bases is in some way arbitrary. The repetitive motif UCAG separates bases according to size (pyrimidine bases U and C, purine bases A and G fall together), groups the inosine-binding bases U, C and A together and allows for the amino acids methionine and isoleucine to be listed in separate groups. When adhering to a constant string of bases, the UCAG motif offers a higher degree of packing of the code as demonstrated by Serguei Lenski on his homepage, which contains a mathematical approach to packing of the genetic code. His results indicate that listing codons in the order of 2-1-3 and retaining the UCAG motif offers an optimal amount of packing, but that does not mean that there aren't any other ways to represent the code. The physicist Yurij Rumer (1901-1985) preferred the motif CGUA for various reasons, placing more emphasis on the strength of bonds formed between the cognate bases (C-G forming three hydrogen bonds, A-U forming only two, see D. A. Semenov's paper if you have no access Rumer's original publication in Russian: Rumer IuB. [Codon systematization in the genetic code] Dokl Akad Nauk SSSR. 1966 Apr 21;167(6):1393-4.).
I believe that the motif AGCU (or UCGA) when viewed from a circular perspective offers a happy compromise between the two. Furthermore, by placing the second codon base at the centre of the diagram it becomes possible to unite the codons of leucine, serine, arginine and stop and to see codons cluster into groups according to the chemical properties of the respective amino acid: M, I, V, L, F are all hydrophobic; K, N, D, E, Q, H, Y are all hydrophilic, T, S and hydoxy-proline carry hydroxyl groups whilst A is structurally related to S; the last group comprises amino acids at the extremes from the smallest G to the largest W, the most hydrophilic R to the hydrophobe C, however S is structurally related to C and C, U, S and G can be converted into one another biosynthetically. In addition, S is a substrate for W synthesis (for more information on the role of biosynthetic pathways in shaping the genetic code, I would recommend: Wong JT. A co-evolution theory of the genetic code. Proc Natl Acad Sci U S A. 1975 May;72(5):1909-12.). Finally, the rare genetically encoded amino acid pyrrolysine (O) found in members of the Methanosarcinaceae family of archeaea is a lysine derivative contianing a pyrrol ring, which reminds me of the structures found in the amino acids W, Y and H and somehow fits neatly into the scheme with proximity to K, H, Y and W.

The 3D model of the genetic code along 2-1-3 rules allows a number of projections that represent distorted "maps" of the code. In this post are three projections of the standard code as well as four diagrams for variants of the standard code, found in mitochondria, mycoplasma, cilliates and green algae, although these graphs are oversimplified. For the recently evolved genetic code in Candida where an L codon has changed into an S codon it is not possible to combine the codons in a way that allows for all codons to be grouped together according to their respective amino acid. Even so, one can follow the small changes that make such a big difference, with the bases 1 and 2 both remaining pyrimidines in the genetic code of Candida. For details of these and more variants of the standard code I would like to draw your attention to the taxonomy browser at NCBI. For more of my thoughts on the genetic code you can follow this link to blog.rna-game.org. Thank you.
Standard code: 1

Standard code: 2

Standard code: 3

Vertebrate Mitochondria Code

Invertebrate Mitochondria Code

Yeast Mitochonria Code
(Codons CGA and CGC absent)

Mycoplasma

Cilliates and green algae

Cilliates

Thursday, August 23, 2007
Another model
When I set out to do realign the genetic code, it was almost like playing a game of sudoku. The intention was to see if there was a way to simplify the way the code was represented as an answer to some who claimed that the code was too complex to have evolved on its own. Now that it looks so shockingly simple, some might claim that it is so perfectly simple that only a designer could have made it. This I cannot agree with, it's not perfect whatever that may mean, but simple physical and chemical forces appear to sufficiently explain the evolution of the code based on the material that was available at the time. There are countless little and big influences that made me have a go at the code, but Tetris and Calder are sure to have played a role in this...
I would suggest that a three-dimensional representation would be an even better model. All you need if you want to build a model yourself at home is a couple of molecular building block kits. Make sure that you have blocks that allow for tetra- and penta-valent binding (e.g. carbon atoms in normal and transitional state).
Start with a tetravalent-binding block at the centre to get the tetrahedrical base, use the binding elements to represent A, G, C, and U and continue with the pentavalent-binding blocks until you reach the level of amino acids where you then can use different coloured blocks to symbolise the individual amino acids.
This approach lets you bring amino acid codons that seemed at opposite poles into close proximity (e.g. K and R or F and Y). I know, it may look confusing to begin with, but it adds another layer of information. The categories for grouping the amino acid codons on www.rna-game.org are after all somewhat subjective (non-polar, transitional, special, polar), maybe there are other categories that should be used to group the codons.
I guess what I mean to say is, have fun. Of course sincerety and truth remain the essence of science and empirical and rational thinking are the backbone of this line of work, but intuition and a sense of wonder are crucial elements as well.

The graphs on www.rna-game.org and blog.rna-game.org are distorted two-dimensional maps of the genetic code in three dimensions as shown above.

I would suggest that a three-dimensional representation would be an even better model. All you need if you want to build a model yourself at home is a couple of molecular building block kits. Make sure that you have blocks that allow for tetra- and penta-valent binding (e.g. carbon atoms in normal and transitional state).
Start with a tetravalent-binding block at the centre to get the tetrahedrical base, use the binding elements to represent A, G, C, and U and continue with the pentavalent-binding blocks until you reach the level of amino acids where you then can use different coloured blocks to symbolise the individual amino acids.
This approach lets you bring amino acid codons that seemed at opposite poles into close proximity (e.g. K and R or F and Y). I know, it may look confusing to begin with, but it adds another layer of information. The categories for grouping the amino acid codons on www.rna-game.org are after all somewhat subjective (non-polar, transitional, special, polar), maybe there are other categories that should be used to group the codons.
I guess what I mean to say is, have fun. Of course sincerety and truth remain the essence of science and empirical and rational thinking are the backbone of this line of work, but intuition and a sense of wonder are crucial elements as well.

The graphs on www.rna-game.org and blog.rna-game.org are distorted two-dimensional maps of the genetic code in three dimensions as shown above.

Saturday, August 18, 2007
Leishmania, inbred strains of mice, SOCS and DNA hypermethylation
While most strains of inbred mice are able to control and largely eliminate parasitic L. major, BALB/c and DBA/2 mice develop non-healing lesions [1]. It has been clearly established that Th1-type cytokines such as IFN‑γ offer protection to the intracellular parasite, while Th2-type cytokines such as IL‑4 are linked to susceptibility [2-5]. Macrophages play a dual role in Leishmania infection because they are part of the innate immune system but also are the primary host cells for the parasite. It has been demonstrated that IFN‑γ drives macrophages to acquire the classically activated phenotype characterised by iNOS expression in murine cells, whereas IL‑4 stimulation of macrophages is recognised to drive them to an alternative state of activation typically expressing the enzyme arginase1 [6-8]. The activity of arginase1 has been correlated to parasite proliferation [9;10]. Arginase breaks down arginine to produce urea and ornithine, which then can be metabolised further to proline (a possible source of energy and a building block for protein synthesis in parasites), polyamines (DNA packaging) and possibly nucleotide biosynthesis (ornithine to glutamine to purine and/or ornithine via ornithine transcarbamoylase to pyrimidine bases?), all of which could or do act as nutrients for the proliferating parasite.
In mice, iNOS plays a very important role in controlling the disease (humans have evolved other mechanisms of Th1 mediation, e.g. Vitamin D receptor-mediated mechanisms [11], which seem to be easily available to sun-exposed humans compared to nocturnal mice). Normally resistant mice that have been made deficient in iNOS become susceptible to L. major, while phox-deficient mice are able to control the disease, implying different modes of action by reactive oxygen species (ROS) generated by phox and reactive nitrogen intermediates (RNI) such as NO [12]. iNOS competes with arginase for the same substrate, arginine. But unlike arginase, iNOS produces citrulline and the radical NO which is not further metabolised to produce a source of energy, building blocks or nucleotide synthesis. In fact, a complex inter-regulatory network of competition and inhibition exists between arginase and iNOS on an enzymatic level (iNOS produces NOHA in the first step to NO catalysis, which acts as an inhibitor to arginase1 [13]) as well as on the complex cross-regulation of cytokines that induce these enzymes (reviewed elsewhere and in [14-16]).
The balance between these two enzymes is therefore of crucial importance to the parasite, with the activity leading to proliferation and the other to parasite killing.
SOCS
SOCS proteins are a relatively recently discovered group of at least eight intracellular proteins that act as negative regulators of cell signalling [17-19]. Their SH2 domain binds to phosphorylated Tyrosine (pY) residues and disrupts cell signalling involving Tyrosine kinases, while their SOCS box-domain has been shown to have ubiquitin E3 ligase activity [20-24]. One major route for poly-ubiquitinated cytosolic proteins is their degradation by the proteasome while mono- or multi-ubiquitination of membrane proteins leads to the internalisation or even lysosomal degradation of these proteins [25]. (see: "http://www.nature.com/nri/journal/v2/n6/full/nri818.html")
Many major cytokine receptors such as IFN‑γR, and IL‑4R do not have an inherent Tyrosine kinase activity themselves, but ligand-induced dimerisation leads to recruitment and activation of Tyrosine kinases of the JAK family which in turn lead to the phosphorylation and activation of STAT transcription factors [19]. Tyrosine phosphorylation is a widely used messaging system for short-lived, reversible and regulated signal transduction. In addition to cytokines such as IFN‑γ and IL‑4, many other receptors use phosphorylation of Tyrosine residues as a signal. They either act directly as Tyrosine kinases themselves, (the insulin receptor phosphorylating itself and its substrates IRS1 and IRS2 see review by Youngren accepted for publication in CMLS 2007), or recruit Tyrosine kinases to phosphorylate downstream signalling adapters for them. Phosphorylation of proteins can also be used to facilitate the recruitment of individual components into larger signalling complexes held together and assembled in the correct order by pY – SH2 domain binding (for instance, it may be possible that phosphorylation of the TLR leads to its recruitment into complexes involving other signalling molecules such as PI3 kinases, adenylate cyclases or G-proteins or other proteins with kinase or lipase activity, not to mention other substrates [26;27]).
Because they are induced by cytokines such as IL‑4 and IFN‑γ, it should come as no surprise that SOCS proteins are differentially expressed in different types of T helper cells, as indeed they are, playing an important regulatory role in the development of different cell types [35].
Mice




The mechanism of action is not entirely clear although or (paradoxically perhaps) because antimony had been used as a medicine since antiquity and in its modern preparation since the first half of the 20th century. Based on observations in responses to helminthic infections, it had been speculated that antimony could interfere with the sugar metabolism of the parasite [74], but promastigotes do seem to be able to survive culture in the presence of antimony. Apart from applications in anti-leishmanial chemotherapy very little is know about the pharmacological properties of antimony. However, arsenic, its smaller and more famous cousin, has been used in a number of pharmacological studies. It ranks highly in the lists of toxic agents and chronic low-dose arsenic exposure has been linked to a number of cancers. However, the mechanism of carcinogenesis is not fully understood yet. It is a known carcinogen yet it is not a potent mutagen in itself. The lack of any prominent signal transduction pathway and animal model for arsenic carcinogenesis has led to the belief that it acts as an epigenetic carcinogen. It was only recently shown that long-term low-dose exposure of arsenic leads to a reduction in intracellular S-adenosylmethionine (SAM) and a loss of DNA methylation [75]. This is most likely due to methylation of arsenic depleting intracellular levels of SAM and an observed repression of DNA methyltransferases DNMT1 and DNMT3A gene expression. (see: "www.ehponline.org/members/2005/8600/8600.html" and "www-ermm.cbcu.cam.ac.uk")
Model
SOCS proteins act as negative regulators to a great number of cell signalling processes including IL‑4R and insulin receptor signalling. The observed correlation between resistance to Leishmania infections (advantageous to the mouse) and insulin resistance (pathologic) in different strains of mice seems to suggest SOCS as the common link. Not only do they inhibit physical association of receptor and adapter proteins by occupying crucial pY residues, their SOCS box domains can act as ubiquitin-E3-ligases promoting the mono-, multi- or poly-ubiquitination of target proteins, resulting in down-regulation of trans-membrane receptors through internalisation, redistribution of membrane bound proteins to endosomal/lysosomal compartments (TLR [78]) or proteasomal degradation of cytosolic proteins (possibly STAT, other SOCS proteins [79]). The SOCS-induced shuttling of TLR to the endosomal/lysosomal pathway could be used in antigen-presenting cells as an efficient way to redirect microbial material to cellular compartments that then ensure preferential/efficient antigen presentation (as demonstrated in the case of Toxoplasma gondii and TLR11 [78]).
E) Is SAM content/ DNA methylation linked to pathogenesis of Leishmania in visceral leishmaniases?
In mice, iNOS plays a very important role in controlling the disease (humans have evolved other mechanisms of Th1 mediation, e.g. Vitamin D receptor-mediated mechanisms [11], which seem to be easily available to sun-exposed humans compared to nocturnal mice). Normally resistant mice that have been made deficient in iNOS become susceptible to L. major, while phox-deficient mice are able to control the disease, implying different modes of action by reactive oxygen species (ROS) generated by phox and reactive nitrogen intermediates (RNI) such as NO [12]. iNOS competes with arginase for the same substrate, arginine. But unlike arginase, iNOS produces citrulline and the radical NO which is not further metabolised to produce a source of energy, building blocks or nucleotide synthesis. In fact, a complex inter-regulatory network of competition and inhibition exists between arginase and iNOS on an enzymatic level (iNOS produces NOHA in the first step to NO catalysis, which acts as an inhibitor to arginase1 [13]) as well as on the complex cross-regulation of cytokines that induce these enzymes (reviewed elsewhere and in [14-16]).
The balance between these two enzymes is therefore of crucial importance to the parasite, with the activity leading to proliferation and the other to parasite killing.
SOCS
SOCS proteins are a relatively recently discovered group of at least eight intracellular proteins that act as negative regulators of cell signalling [17-19]. Their SH2 domain binds to phosphorylated Tyrosine (pY) residues and disrupts cell signalling involving Tyrosine kinases, while their SOCS box-domain has been shown to have ubiquitin E3 ligase activity [20-24]. One major route for poly-ubiquitinated cytosolic proteins is their degradation by the proteasome while mono- or multi-ubiquitination of membrane proteins leads to the internalisation or even lysosomal degradation of these proteins [25]. (see: "http://www.nature.com/nri/journal/v2/n6/full/nri818.html")
Many major cytokine receptors such as IFN‑γR, and IL‑4R do not have an inherent Tyrosine kinase activity themselves, but ligand-induced dimerisation leads to recruitment and activation of Tyrosine kinases of the JAK family which in turn lead to the phosphorylation and activation of STAT transcription factors [19]. Tyrosine phosphorylation is a widely used messaging system for short-lived, reversible and regulated signal transduction. In addition to cytokines such as IFN‑γ and IL‑4, many other receptors use phosphorylation of Tyrosine residues as a signal. They either act directly as Tyrosine kinases themselves, (the insulin receptor phosphorylating itself and its substrates IRS1 and IRS2 see review by Youngren accepted for publication in CMLS 2007), or recruit Tyrosine kinases to phosphorylate downstream signalling adapters for them. Phosphorylation of proteins can also be used to facilitate the recruitment of individual components into larger signalling complexes held together and assembled in the correct order by pY – SH2 domain binding (for instance, it may be possible that phosphorylation of the TLR leads to its recruitment into complexes involving other signalling molecules such as PI3 kinases, adenylate cyclases or G-proteins or other proteins with kinase or lipase activity, not to mention other substrates [26;27]).
Because they are induced by cytokines such as IL‑4 and IFN‑γ, it should come as no surprise that SOCS proteins are differentially expressed in different types of T helper cells, as indeed they are, playing an important regulatory role in the development of different cell types [35].
Mice




The mechanism of action is not entirely clear although or (paradoxically perhaps) because antimony had been used as a medicine since antiquity and in its modern preparation since the first half of the 20th century. Based on observations in responses to helminthic infections, it had been speculated that antimony could interfere with the sugar metabolism of the parasite [74], but promastigotes do seem to be able to survive culture in the presence of antimony. Apart from applications in anti-leishmanial chemotherapy very little is know about the pharmacological properties of antimony. However, arsenic, its smaller and more famous cousin, has been used in a number of pharmacological studies. It ranks highly in the lists of toxic agents and chronic low-dose arsenic exposure has been linked to a number of cancers. However, the mechanism of carcinogenesis is not fully understood yet. It is a known carcinogen yet it is not a potent mutagen in itself. The lack of any prominent signal transduction pathway and animal model for arsenic carcinogenesis has led to the belief that it acts as an epigenetic carcinogen. It was only recently shown that long-term low-dose exposure of arsenic leads to a reduction in intracellular S-adenosylmethionine (SAM) and a loss of DNA methylation [75]. This is most likely due to methylation of arsenic depleting intracellular levels of SAM and an observed repression of DNA methyltransferases DNMT1 and DNMT3A gene expression. (see: "www.ehponline.org/members/2005/8600/8600.html" and "www-ermm.cbcu.cam.ac.uk")
Model
SOCS proteins act as negative regulators to a great number of cell signalling processes including IL‑4R and insulin receptor signalling. The observed correlation between resistance to Leishmania infections (advantageous to the mouse) and insulin resistance (pathologic) in different strains of mice seems to suggest SOCS as the common link. Not only do they inhibit physical association of receptor and adapter proteins by occupying crucial pY residues, their SOCS box domains can act as ubiquitin-E3-ligases promoting the mono-, multi- or poly-ubiquitination of target proteins, resulting in down-regulation of trans-membrane receptors through internalisation, redistribution of membrane bound proteins to endosomal/lysosomal compartments (TLR [78]) or proteasomal degradation of cytosolic proteins (possibly STAT, other SOCS proteins [79]). The SOCS-induced shuttling of TLR to the endosomal/lysosomal pathway could be used in antigen-presenting cells as an efficient way to redirect microbial material to cellular compartments that then ensure preferential/efficient antigen presentation (as demonstrated in the case of Toxoplasma gondii and TLR11 [78]).
E) Is SAM content/ DNA methylation linked to pathogenesis of Leishmania in visceral leishmaniases?
References
1. Sacks,D. and Noben-Trauth,N., The immunology of susceptibility and resistance to Leishmania major in mice. Nat.Rev.Immunol. 2002. 2: 845-858.
2. Noben-Trauth,N., Kropf,P., and Muller,I., Susceptibility to Leishmania major infection in interleukin-4-deficient mice. Science 1996. 271: 987-990.
3. Noben-Trauth,N., Paul,W.E., and Sacks,D.L., IL-4- and IL-4 receptor-deficient BALB/c mice reveal differences in susceptibility to Leishmania major parasite substrains. J.Immunol. 1999. 162: 6132-6140.
4. Noben-Trauth,N., Susceptibility to Leishmania major infection in the absence of IL-4. Immunol.Lett. 2000. 75: 41-44.
5. Noben-Trauth,N., Lira,R., Nagase,H., Paul,W.E., and Sacks,D.L., The relative contribution of IL-4 receptor signaling and IL-10 to susceptibility to Leishmania major. J.Immunol. 2003. 170: 5152-5158.
6. Gordon,S., Alternative activation of macrophages. Nat.Rev.Immunol. 2003. 3: 23-35.
7. Hesse,M., Modolell,M., La Flamme,A.C., Schito,M., Fuentes,J.M., Cheever,A.W., Pearce,E.J., and Wynn,T.A., Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J.Immunol. 2001. 167: 6533-6544.
8. Modolell,M., Corraliza,I.M., Link,F., Soler,G., and Eichmann,K., Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur.J.Immunol. 1995. 25: 1101-1104.
9. Kropf,P., Fuentes,J.M., Fahnrich,E., Arpa,L., Herath,S., Weber,V., Soler,G., Celada,A., Modolell,M., and Muller,I., Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. FASEB J. 2005. 19: 1000-1002.
10. Corraliza,I.M., Soler,G., Eichmann,K., and Modolell,M., Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem.Biophys.Res.Commun. 1995. 206: 667-673.
11. Liu,P.T., Stenger,S., Li,H., Wenzel,L., Tan,B.H., Krutzik,S.R., Ochoa,M.T., Schauber,J., Wu,K., Meinken,C., Kamen,D.L., Wagner,M., Bals,R., Steinmeyer,A., Zugel,U., Gallo,R.L., Eisenberg,D., Hewison,M., Hollis,B.W., Adams,J.S., Bloom,B.R., and Modlin,R.L., Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006. 311: 1770-1773.
12. Murray,H.W. and Nathan,C.F., Macrophage microbicidal mechanisms in vivo: reactive nitrogen versus oxygen intermediates in the killing of intracellular visceral Leishmania donovani. J.Exp.Med. 1999. 189: 741-746.
13. Tenu,J.P., Lepoivre,M., Moali,C., Brollo,M., Mansuy,D., and Boucher,J.L., Effects of the new arginase inhibitor N(omega)-hydroxy-nor-L-arginine on NO synthase activity in murine macrophages. Nitric.Oxide. 1999. 3: 427-438.
14. Cher,D.J. and Mosmann,T.R., Two types of murine helper T cell clone. II. Delayed-type hypersensitivity is mediated by TH1 clones. J.Immunol. 1987. 138: 3688-3694.
15. Mosmann,T.R., Cherwinski,H., Bond,M.W., Giedlin,M.A., and Coffman,R.L., Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J.Immunol. 1986. 136: 2348-2357.
16. Gor,D.O., Rose,N.R., and Greenspan,N.S., TH1-TH2: a procrustean paradigm. Nat.Immunol. 2003. 4: 503-505.
17. Alexander,W.S., Suppressors of cytokine signalling (SOCS) in the immune system. Nat.Rev.Immunol. 2002. 2: 410-416.
18. Alexander,W.S. and Hilton,D.J., The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu.Rev.Immunol. 2004. 22: 503-529.
19. O'sullivan,L.A., Liongue,C., Lewis,R.S., Stephenson,S.E., and Ward,A.C., Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol.Immunol. 2007.
20. Rui,L., Yuan,M., Frantz,D., Shoelson,S., and White,M.F., SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J.Biol.Chem. 2002. 277: 42394-42398.
21. Kile,B.T., Schulman,B.A., Alexander,W.S., Nicola,N.A., Martin,H.M., and Hilton,D.J., The SOCS box: a tale of destruction and degradation. Trends Biochem.Sci. 2002. 27: 235-241.
22. Krebs,D.L., Uren,R.T., Metcalf,D., Rakar,S., Zhang,J.G., Starr,R., De Souza,D.P., Hanzinikolas,K., Eyles,J., Connolly,L.M., Simpson,R.J., Nicola,N.A., Nicholson,S.E., Baca,M., Hilton,D.J., and Alexander,W.S., SOCS-6 binds to insulin receptor substrate 4, and mice lacking the SOCS-6 gene exhibit mild growth retardation. Mol.Cell Biol. 2002. 22: 4567-4578.
23. Zhang,J.G., Metcalf,D., Rakar,S., Asimakis,M., Greenhalgh,C.J., Willson,T.A., Starr,R., Nicholson,S.E., Carter,W., Alexander,W.S., Hilton,D.J., and Nicola,N.A., The SOCS box of suppressor of cytokine signaling-1 is important for inhibition of cytokine action in vivo. Proc.Natl.Acad.Sci.U.S.A 2001. 98: 13261-13265.
24. Kamizono,S., Hanada,T., Yasukawa,H., Minoguchi,S., Kato,R., Minoguchi,M., Hattori,K., Hatakeyama,S., Yada,M., Morita,S., Kitamura,T., Kato,H., Nakayama,K., and Yoshimura,A., The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J.Biol.Chem. 2001. 276: 12530-12538.
25. Mukhopadhyay,D. and Riezman,H., Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007. 315: 201-205.
26. Hajishengallis,G., Tapping,R.I., Harokopakis,E., Nishiyama,S., Ratti,P., Schifferle,R.E., Lyle,E.A., Triantafilou,M., Triantafilou,K., and Yoshimura,F., Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. 2006. 8: 1557-1570.
27. Triantafilou,M., Gamper,F.G., Haston,R.M., Mouratis,M.A., Morath,S., Hartung,T., and Triantafilou,K., Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J.Biol.Chem. 2006. 281: 31002-31011.
28. Dickensheets,H., Vazquez,N., Sheikh,F., Gingras,S., Murray,P.J., Ryan,J.J., and Donnelly,R.P., Suppressor of cytokine signaling-1 is an IL-4-inducible gene in macrophages and feedback inhibits IL-4 signaling. Genes Immun. 2007. 8: 21-27.
29. Chong,M.M., Chen,Y., Darwiche,R., Dudek,N.L., Irawaty,W., Santamaria,P., Allison,J., Kay,T.W., and Thomas,H.E., Suppressor of cytokine signaling-1 overexpression protects pancreatic beta cells from CD8+ T cell-mediated autoimmune destruction. J.Immunol. 2004. 172: 5714-5721.
30. Ueki,K., Kondo,T., and Kahn,C.R., Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol.Cell Biol. 2004. 24: 5434-5446.
31. Starr,R., Metcalf,D., Elefanty,A.G., Brysha,M., Willson,T.A., Nicola,N.A., Hilton,D.J., and Alexander,W.S., Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc.Natl.Acad.Sci.U.S.A 1998. 95: 14395-14399.
32. Marine,J.C., Topham,D.J., McKay,C., Wang,D., Parganas,E., Stravopodis,D., Yoshimura,A., and Ihle,J.N., SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell 1999. 98: 609-616.
33. Alexander,W.S., Starr,R., Fenner,J.E., Scott,C.L., Handman,E., Sprigg,N.S., Corbin,J.E., Cornish,A.L., Darwiche,R., Owczarek,C.M., Kay,T.W., Nicola,N.A., Hertzog,P.J., Metcalf,D., and Hilton,D.J., SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 1999. 98: 597-608.
34. Roberts,A.W., Robb,L., Rakar,S., Hartley,L., Cluse,L., Nicola,N.A., Metcalf,D., Hilton,D.J., and Alexander,W.S., Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc.Natl.Acad.Sci.U.S.A 2001. 98: 9324-9329.
35. Inoue,H. and Kubo,M., SOCS proteins in T helper cell differentiation: implications for allergic disorders? Expert.Rev.Mol.Med. 2004. 6: 1-11.
36. Baetz,A., Frey,M., Heeg,K., and Dalpke,A.H., Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J.Biol.Chem. 2004. 279: 54708-54715.
37. Nakagawa,R., Naka,T., Tsutsui,H., Fujimoto,M., Kimura,A., Abe,T., Seki,E., Sato,S., Takeuchi,O., Takeda,K., Akira,S., Yamanishi,K., Kawase,I., Nakanishi,K., and Kishimoto,T., SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002. 17: 677-687.
38. Campbell,I.L., Cytokine-mediated inflammation, tumorigenesis, and disease-associated JAK/STAT/SOCS signaling circuits in the CNS. Brain Res.Brain Res.Rev. 2005. 48: 166-177.
39. He,B., You,L., Uematsu,K., Zang,K., Xu,Z., Lee,A.Y., Costello,J.F., McCormick,F., and Jablons,D.M., SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc.Natl.Acad.Sci.U.S.A 2003. 100: 14133-14138.
40. Weber,A., Hengge,U.R., Bardenheuer,W., Tischoff,I., Sommerer,F., Markwarth,A., Dietz,A., Wittekind,C., and Tannapfel,A., SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene 2005. 24: 6699-6708.
41. Evans,M.K., Yu,C.R., Lohani,A., Mahdi,R.M., Liu,X., Trzeciak,A.R., and Egwuagu,C.E., Expression of SOCS1 and SOCS3 genes is differentially regulated in breast cancer cells in response to proinflammatory cytokine and growth factor signals. Oncogene 2006.
42. Bellezza,I., Neuwirt,H., Nemes,C., Cavarretta,I.T., Puhr,M., Steiner,H., Minelli,A., Bartsch,G., Offner,F., Hobisch,A., Doppler,W., and Culig,Z., Suppressor of cytokine signaling-3 antagonizes cAMP effects on proliferation and apoptosis and is expressed in human prostate cancer. Am.J.Pathol. 2006. 169: 2199-2208.
43. Trojan,L., Schaaf,A., Steidler,A., Haak,M., Thalmann,G., Knoll,T., Gretz,N., Alken,P., and Michel,M.S., Identification of metastasis-associated genes in prostate cancer by genetic profiling of human prostate cancer cell lines. Anticancer Res. 2005. 25: 183-191.
44. Somasundar,P., Frankenberry,K.A., Skinner,H., Vedula,G., McFadden,D.W., Riggs,D., Jackson,B., Vangilder,R., Hileman,S.M., and Vona-Davis,L.C., Prostate cancer cell proliferation is influenced by leptin. J.Surg.Res. 2004. 118: 71-82.
45. Heinrich,P.C., Behrmann,I., Haan,S., Hermanns,H.M., Muller-Newen,G., and Schaper,F., Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem.J. 2003. 374: 1-20.
46. He,B., You,L., Xu,Z., Mazieres,J., Lee,A.Y., and Jablons,D.M., Activity of the suppressor of cytokine signaling-3 promoter in human non-small-cell lung cancer. Clin.Lung Cancer 2004. 5: 366-370.
47. He,B., You,L., Uematsu,K., Zang,K., Xu,Z., Lee,A.Y., Costello,J.F., McCormick,F., and Jablons,D.M., SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc.Natl.Acad.Sci.U.S.A 2003. 100: 14133-14138.
48. Kafetzis,D.A., An overview of paediatric leishmaniasis. J.Postgrad.Med. 2003. 49: 31-38.
49. Laakso,M., Insulin resistance and its impact on the approach to therapy of type 2 diabetes. Int.J.Clin.Pract.Suppl 2001. 8-12.
50. Laakso,M., Insulin resistance and coronary heart disease. Curr.Opin.Lipidol. 1996. 7: 217-226.
51. Tortorella,C., Simone,O., Piazzolla,G., Stella,I., Cappiello,V., and Antonaci,S., Role of phosphoinositide 3-kinase and extracellular signal-regulated kinase pathways in granulocyte macrophage-colony-stimulating factor failure to delay fas-induced neutrophil apoptosis in elderly humans. J.Gerontol.A Biol.Sci.Med.Sci. 2006. 61: 1111-1118.
52. Tortorella,C., Simone,O., Piazzolla,G., Stella,I., and Antonaci,S., Age-related impairment of GM-CSF-induced signalling in neutrophils: Role of SHP-1 and SOCS proteins. Ageing Res.Rev. 2006.
53. Tortorella,C., Stella,I., Piazzolla,G., Cappiello,V., Simone,O., Pisconti,A., and Antonaci,S., Impaired interleukin-12-dependent T-cell functions during aging: role of signal transducer and activator of transcription 4 (STAT4) and suppressor of cytokine signaling 3 (SOCS3). J.Gerontol.A Biol.Sci.Med.Sci. 2006. 61: 125-135.
54. Peralta,S., Carrascosa,J.M., Gallardo,N., Ros,M., and Arribas,C., Ageing increases SOCS-3 expression in rat hypothalamus: effects of food restriction. Biochem.Biophys.Res.Commun. 2002. 296: 425-428.
55. Welch,J.S., Escoubet-Lozach,L., Sykes,D.B., Liddiard,K., Greaves,D.R., and Glass,C.K., TH2 cytokines and allergic challenge induce Ym1 expression in macrophages by a STAT6-dependent mechanism. J.Biol.Chem. 2002. 277: 42821-42829.
56. Julia,V. and Glaichenhaus,N., CD4(+) T cells which react to the Leishmania major LACK antigen rapidly secrete interleukin-4 and are detrimental to the host in resistant B10.D2 mice. Infect.Immun. 1999. 67: 3641-3644.
57. Launois,P., Maillard,I., Pingel,S., Swihart,K.G., Xenarios,I., cha-Orbea,H., Diggelmann,H., Locksley,R.M., MacDonald,H.R., and Louis,J.A., IL-4 rapidly produced by V beta 4 V alpha 8 CD4+ T cells instructs Th2 development and susceptibility to Leishmania major in BALB/c mice. Immunity. 1997. 6: 541-549.
58. Maillard,I., Launois,P., Himmelrich,H., cha-Orbea,H., Diggelmann,H., Locksley,R.M., and Louis,J.A., Functional plasticity of the LACK-reactive Vbeta4-Valpha8 CD4(+) T cells normally producing the early IL-4 instructing Th2 cell development and susceptibility to Leishmania major in BALB / c mice. Eur.J.Immunol. 2001. 31: 1288-1296.
59. Julia,V., McSorley,S.S., Malherbe,L., Breittmayer,J.P., Girard-Pipau,F., Beck,A., and Glaichenhaus,N., Priming by microbial antigens from the intestinal flora determines the ability of CD4+ T cells to rapidly secrete IL-4 in BALB/c mice infected with Leishmania major. J.Immunol. 2000. 165: 5637-5645.
60. Julia,V., Rassoulzadegan,M., and Glaichenhaus,N., Resistance to Leishmania major induced by tolerance to a single antigen. Science 1996. 274: 421-423.
61. Scott,P., Eaton,A., Gause,W.C., di,Z., X, and Hondowicz,B., Early IL-4 production does not predict susceptibility to Leishmania major. Exp.Parasitol. 1996. 84: 178-187.
62. Graham,A.L., Taylor,M.D., Le,G.L., Lamb,T.J., Magennis,M., and Allen,J.E., Quantitative appraisal of murine filariasis confirms host strain differences but reveals that BALB/c females are more susceptible than males to Litomosoides sigmodontis. Microbes.Infect. 2005. 7: 612-618.
63. Le,G.L., Lamb,T.J., Graham,A.L., Harcus,Y., and Allen,J.E., IL-4 is required to prevent filarial nematode development in resistant but not susceptible strains of mice. Int.J.Parasitol. 2002. 32: 1277-1284.
64. de Veer,M.J., Curtis,J.M., Baldwin,T.M., DiDonato,J.A., Sexton,A., McConville,M.J., Handman,E., and Schofield,L., MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur.J.Immunol. 2003. 33: 2822-2831.
65. Flandin,J.F., Chano,F., and Descoteaux,A., RNA interference reveals a role for TLR2 and TLR3 in the recognition of Leishmania donovani promastigotes by interferon-gamma-primed macrophages. Eur.J.Immunol. 2006. 36: 411-420.
66. Kropf,P., Freudenberg,M.A., Modolell,M., Price,H.P., Herath,S., Antoniazi,S., Galanos,C., Smith,D.F., and Muller,I., Toll-like receptor 4 contributes to efficient control of infection with the protozoan parasite Leishmania major. Infect.Immun. 2004. 72: 1920-1928.
67. Debus,A., Glasner,J., Rollinghoff,M., and Gessner,A., High levels of susceptibility and T helper 2 response in MyD88-deficient mice infected with Leishmania major are interleukin-4 dependent. Infect.Immun. 2003. 71: 7215-7218.
68. Muraille,E., De,T.C., Brait,M., De,B.P., Leo,O., and Carlier,Y., Genetically resistant mice lacking MyD88-adapter protein display a high susceptibility to Leishmania major infection associated with a polarized Th2 response. J.Immunol. 2003. 170: 4237-4241.
69. Carriere,V., Roussel,L., Ortega,N., Lacorre,D.A., Americh,L., Aguilar,L., Bouche,G., and Girard,J.P., IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc.Natl.Acad.Sci.U.S.A 2007. 104: 282-287.
70. Schmitz,J., Owyang,A., Oldham,E., Song,Y., Murphy,E., McClanahan,T.K., Zurawski,G., Moshrefi,M., Qin,J., Li,X., Gorman,D.M., Bazan,J.F., and Kastelein,R.A., IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005. 23: 479-490.
71. Dinarello,C.A., An IL-1 family member requires caspase-1 processing and signals through the ST2 receptor. Immunity. 2005. 23: 461-462.
72. Takezako,N., Hayakawa,M., Hayakawa,H., Aoki,S., Yanagisawa,K., Endo,H., and Tominaga,S., ST2 suppresses IL-6 production via the inhibition of IkappaB degradation induced by the LPS signal in THP-1 cells. Biochem.Biophys.Res.Commun. 2006. 341: 425-432.
73. Sweet,M.J., Leung,B.P., Kang,D., Sogaard,M., Schulz,K., Trajkovic,V., Campbell,C.C., Xu,D., and Liew,F.Y., A novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J.Immunol. 2001. 166: 6633-6639.
74. Singh,N., Drug resistance mechanisms in clinical isolates of Leishmania donovani. Indian J.Med.Res. 2006. 123: 411-422.
75. Reichard,J.F., Schnekenburger,M., and Puga,A., Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem.Biophys.Res.Commun. 2007. 352: 188-192.
76. Bullen,D.V., Baldwin,T.M., Curtis,J.M., Alexander,W.S., and Handman,E., Persistence of lesions in suppressor of cytokine signaling-1-deficient mice infected with Leishmania major. J.Immunol. 2003. 170: 4267-4272.
77. Bertholet,S., Dickensheets,H.L., Sheikh,F., Gam,A.A., Donnelly,R.P., and Kenney,R.T., Leishmania donovani-induced expression of suppressor of cytokine signaling 3 in human macrophages: a novel mechanism for intracellular parasite suppression of activation. Infect.Immun. 2003. 71: 2095-2101.
78. Yarovinsky,F., Kanzler,H., Hieny,S., Coffman,R.L., and Sher,A., Toll-like receptor recognition regulates immunodominance in an antimicrobial CD4+ T cell response. Immunity. 2006. 25: 655-664.
79. Tannahill,G.M., Elliott,J., Barry,A.C., Hibbert,L., Cacalano,N.A., and Johnston,J.A., SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol.Cell Biol. 2005. 25: 9115-9126.
80. Chan,M.W., Chu,E.S., To,K.F., and Leung,W.K., Quantitative detection of methylated SOCS-1 , a tumor suppressor gene, by a modified protocol of quantitative real time methylation-specific PCR using SYBR green and its use in early gastric cancer detection. Biotechnol.Lett. 2004. 26: 1289-1293.
81. Liu,Z., Sanchez,M.A., Jiang,X., Boles,E., Landfear,S.M., and Rosen,B.P., Mammalian glucose permease GLUT1 facilitates transport of arsenic trioxide and methylarsonous acid. Biochem.Biophys.Res.Commun. 2006. 351: 424-430.
Subscribe to:
Posts (Atom)